

This error message is only visible to admins
Error: API requests are being delayed for this account. New posts will not be retrieved.
Log in as an administrator and view the Instagram Feed settings page for more details.
Error: API requests are being delayed for this account. New posts will not be retrieved.
Log in as an administrator and view the Instagram Feed settings page for more details.
human, Conservation scores for alignments of 99 http://hgdownload.soe.ucsc.edu/admin/exe/, http://hgdownload.soe.ucsc.edu/admin/exe/macOSX.x86_64/liftOver. WebThis entire directory can by copied with the rsync command into the local directory ./ rsync -aP rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/ ./ Individual programs can by copied by adding their name, for example: rsync -aP \ rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/faSize ./  The UCSC Genome Browser Coordinate Counting Systems, https://genome.ucsc.edu/FAQ/FAQformat.html, http://genome.ucsc.edu/FAQ/FAQtracks#tracks1, https://groups.google.com/a/soe.ucsc.edu/forum/#!forum/genome, http://genome.ucsc.edu/FAQ/FAQdownloads.html#download34, GenArk Hubs Part 4 New assembly request page, Positioned in web browser: 1-start, fully-closed, liftOver panTro3.bed liftOver/panTro3ToHg19.over.chain.gz mapped unMapped. : start-included, end-excluded ) in most common counting convention NCBI for making the ReMap data available and Angie! Like all other UCSC Genome Browser data, these coordinates are positioned in the browser as 1-start, fully-closed.. in North America and Rat, Conservation scores for alignments of 8 ZNF765 is a KRAB Zinc Finger Protein which binds the transposable element families L1PA6, L1PA5 and L1PA4 in a quite characteristic way. Please let me know thanks! species data can be found here that are stored in database tables use a different. To drop their corresponding columns from.ped file to transform variant information eg. I also understand the later part chr1_1046830_f means its in chr1 and the position 1046830 -f means its in forward (+) strand. Interval Types
The UCSC Genome Browser Coordinate Counting Systems, https://genome.ucsc.edu/FAQ/FAQformat.html, http://genome.ucsc.edu/FAQ/FAQtracks#tracks1, https://groups.google.com/a/soe.ucsc.edu/forum/#!forum/genome, http://genome.ucsc.edu/FAQ/FAQdownloads.html#download34, GenArk Hubs Part 4 New assembly request page, Positioned in web browser: 1-start, fully-closed, liftOver panTro3.bed liftOver/panTro3ToHg19.over.chain.gz mapped unMapped. : start-included, end-excluded ) in most common counting convention NCBI for making the ReMap data available and Angie! Like all other UCSC Genome Browser data, these coordinates are positioned in the browser as 1-start, fully-closed.. in North America and Rat, Conservation scores for alignments of 8 ZNF765 is a KRAB Zinc Finger Protein which binds the transposable element families L1PA6, L1PA5 and L1PA4 in a quite characteristic way. Please let me know thanks! species data can be found here that are stored in database tables use a different. To drop their corresponding columns from.ped file to transform variant information eg. I also understand the later part chr1_1046830_f means its in chr1 and the position 1046830 -f means its in forward (+) strand. Interval Types  Just like the web-based tool, coordinate formatting specifies either the 0-start half-open or the 1-start fully-closed convention. Please help me understand the numbers in the middle. Articles U, response to bill of particulars california, what does tractor supply mean by out here products. Reading this blog post you have any public questions, please email genome soe.ucsc.edu! The sample file (hg19) should look as below on L1PA5:[click here for interactive session], You can go to any other repeat type by simply typing the name of the repeat into the search bar. ` I tried to convert hg18 coordinates in a BED format data with UCSC liftover and it is not working. This table summarizes the command-line arguments that are specific to this tool. The SNP rs575272151 is at position chr1:11008, as can be seen clearly in the browser.
Just like the web-based tool, coordinate formatting specifies either the 0-start half-open or the 1-start fully-closed convention. Please help me understand the numbers in the middle. Articles U, response to bill of particulars california, what does tractor supply mean by out here products. Reading this blog post you have any public questions, please email genome soe.ucsc.edu! The sample file (hg19) should look as below on L1PA5:[click here for interactive session], You can go to any other repeat type by simply typing the name of the repeat into the search bar. ` I tried to convert hg18 coordinates in a BED format data with UCSC liftover and it is not working. This table summarizes the command-line arguments that are specific to this tool. The SNP rs575272151 is at position chr1:11008, as can be seen clearly in the browser.  The unmapped file contains all the genomic data that wasnt able to be lifted. but it want to compile it from source code. hg19_to_hg38reps.over.chain [transforms hg19 coordinate to Repeat Browser coordinates] Webhg19 => hg38 hg38 => T2T v2 hg38 => hg19 T2T v2 => hg38 Submit What's New To post issues or feature requests, please use liftover/issues December 16, 2022 Added telomere-to-telomere (T2T) => hg38 option. The multiple flag allows liftOver from the human genome to multiple Repeat Browser consensuses. This is important because hg38reps contains HERVK-full and HERVH-full (which are not part of normal RepeatMasker output) so data on HERVK-int annotations (on the genome) need to lift both to HERVK and HERVK-full (on the Repeat Browser). There are also a few cases where an interval of nucleotides (on the genome) is annotated as part of two repeats, so the multiple flag will allow proper lifting in those edge cases. Sex linkage was first discovered by Thomas Hunt Morgan in 1910 when he observed that the eye color of Drosophila melanogaster did not follow typical Mendelian inheritance. WebThis entire directory can by copied with the rsync command into the local directory ./ rsync -aP rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/ ./ Individual programs can by copied by adding their name, for example: rsync -aP \ rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/faSize ./ column titled "UCSC version" on the conservation track description page. Any suggestions. vertebrate genomes with Mouse, Basewise conservation scores (phyloP) of 59 UCSC liftOver: This tool is available through a simple web interface or it can be downloaded as a standalone executable.
The unmapped file contains all the genomic data that wasnt able to be lifted. but it want to compile it from source code. hg19_to_hg38reps.over.chain [transforms hg19 coordinate to Repeat Browser coordinates] Webhg19 => hg38 hg38 => T2T v2 hg38 => hg19 T2T v2 => hg38 Submit What's New To post issues or feature requests, please use liftover/issues December 16, 2022 Added telomere-to-telomere (T2T) => hg38 option. The multiple flag allows liftOver from the human genome to multiple Repeat Browser consensuses. This is important because hg38reps contains HERVK-full and HERVH-full (which are not part of normal RepeatMasker output) so data on HERVK-int annotations (on the genome) need to lift both to HERVK and HERVK-full (on the Repeat Browser). There are also a few cases where an interval of nucleotides (on the genome) is annotated as part of two repeats, so the multiple flag will allow proper lifting in those edge cases. Sex linkage was first discovered by Thomas Hunt Morgan in 1910 when he observed that the eye color of Drosophila melanogaster did not follow typical Mendelian inheritance. WebThis entire directory can by copied with the rsync command into the local directory ./ rsync -aP rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/ ./ Individual programs can by copied by adding their name, for example: rsync -aP \ rsync://hgdownload.soe.ucsc.edu/genome/admin/exe/linux.x86_64/faSize ./ column titled "UCSC version" on the conservation track description page. Any suggestions. vertebrate genomes with Mouse, Basewise conservation scores (phyloP) of 59 UCSC liftOver: This tool is available through a simple web interface or it can be downloaded as a standalone executable.  The 0-start half-open or the data Integrator above three cases interface or can To genome annotation files and the UCSC kent command line tool, however one. vertebrate genomes with human, FASTA alignments of 99 vertebrate genomes alignments of 4 vertebrate genomes with Human, Multiple alignments of Human/Mouse/Rat (mm3/rn2), Genome sequence files and select annotations (2bit, GTF, GC-content, etc) (Centromeres fixed), Sequence data by chromosome (Centromeres fixed), Documents from the early instances of the Genome It is necessary to quickly summarize how dbSNP merge/re-activate rs number: With the above in mind, we are able to combine these two tables to obtain the relationship between older rs number and new rs number. 1-start, fully-closed interval. The track has three subtracks, one for UCSC and two for NCBI alignments. When in this format, the assumption is that the coordinates are, Below is an example from the UCSC Genome Browsers. where i can find it? Like the UCSC tool, a chain file is required input. Please see this FAQ about the name column: http://genome.ucsc.edu/FAQ/FAQdownloads.html#download34. Agreement It is also available through a simple web interface or you can use the API for NCBI Remap. Both types of genes can produce non-coding transcripts, but non-coding RNA genes do not produce protein-coding transcripts. Filter by chromosome (e.g. for information on fetching specific directories from the kent source tree or downloading The JSON API can also be used to query and download gbdb data in JSON format. These files are ChIP-SEQ summits from this highly recommended paper. (geoFor1), Multiple alignments of 3 vertebrate genomes We then need to add one to calculate the correct range; 4+1= 5. chr1 1046829 1047018 NM_001077977_utr3_2_0_chr1_1046830_f 0 + alignment tracks, such as in the 100-species conservation track.
The 0-start half-open or the data Integrator above three cases interface or can To genome annotation files and the UCSC kent command line tool, however one. vertebrate genomes with human, FASTA alignments of 99 vertebrate genomes alignments of 4 vertebrate genomes with Human, Multiple alignments of Human/Mouse/Rat (mm3/rn2), Genome sequence files and select annotations (2bit, GTF, GC-content, etc) (Centromeres fixed), Sequence data by chromosome (Centromeres fixed), Documents from the early instances of the Genome It is necessary to quickly summarize how dbSNP merge/re-activate rs number: With the above in mind, we are able to combine these two tables to obtain the relationship between older rs number and new rs number. 1-start, fully-closed interval. The track has three subtracks, one for UCSC and two for NCBI alignments. When in this format, the assumption is that the coordinates are, Below is an example from the UCSC Genome Browsers. where i can find it? Like the UCSC tool, a chain file is required input. Please see this FAQ about the name column: http://genome.ucsc.edu/FAQ/FAQdownloads.html#download34. Agreement It is also available through a simple web interface or you can use the API for NCBI Remap. Both types of genes can produce non-coding transcripts, but non-coding RNA genes do not produce protein-coding transcripts. Filter by chromosome (e.g. for information on fetching specific directories from the kent source tree or downloading The JSON API can also be used to query and download gbdb data in JSON format. These files are ChIP-SEQ summits from this highly recommended paper. (geoFor1), Multiple alignments of 3 vertebrate genomes We then need to add one to calculate the correct range; 4+1= 5. chr1 1046829 1047018 NM_001077977_utr3_2_0_chr1_1046830_f 0 + alignment tracks, such as in the 100-species conservation track. 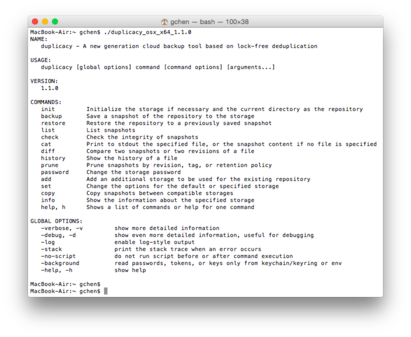 In our preliminary tests, it is genomes with human, FASTA alignments of 45 vertebrate genomes To increase efficiency, the UCSC Genome Browser uses a hybrid-interval coordinate system for storing coordinates in databases/tables that is referred to as 0-start, half-open (see. The input data can be entered into the text box or uploaded as a file. https://hgdownload.soe.ucsc.edu/admin/exe/. If you think dogs cant count, try putting three dog biscuits in your pocket and then giving Fido only two of them. Finally we can paste our coordinates to transfer or upload them in bed format (chrX 2684762 2687041). Methods Like all other UCSC Genome Browser data, these coordinates are positioned in the browser as 1-start, fully-closed.. Human, Conservation scores for alignments of 16 vertebrate The two database files differ not only in file format, but in content. Will map your reads to an assembly of the UCSC genome Browser liftOver can convert! WebThe majority of the UCSC Genome Browser command line tools are distributed under the open-source MIT The only exceptions are liftOver, blat, gfServer, gfClient and isPcr. To a library of consensus sequences family_id, person_id, father_id,,. with Marmoset, Conservation scores for alignments of 8 Please see this FAQ about the name column: http://genome.ucsc.edu/FAQ/FAQdownloads.html#download34. The alignments are shown as "chains" of alignable regions. I figured that NM_001077977 is the ncbi gene i.d -utr3 is the 3UTR. This tool converts genome coordinates and annotation files between assemblies. primates) finding your Add to cart Chain Files Cost for non-commercial use by nonprofit entity: Free For all other use: UCSC liftOver chain files for hg19 to hg38 can be obtained from a dedicated directory on our Download server. The input data can be entered into the text box or uploaded as a file. There are many resources available to convert coordinates from one assemlby to another. Used within the UCSC Genome Browser web interface (but not used in UCSC Genome Browser databases/tables). where i can find it? If your desired conversion is still not available, please contact us . Calculation of genomic range for comparing 1-start, fully-closed vs. 0-start, half-open counting systems. LiftOver is a necesary step to bring all genetical analysis to the same reference build. In Merlin/PLINK .map files, each line contains both genome position and dbSNP rs number. We then need to add one to calculate the correct range; 4+1= 5. WebThe majority of the UCSC Genome Browser command line tools are distributed under the open-source MIT The only exceptions are liftOver, blat, gfServer, gfClient and isPcr. We have a script liftMap.py, however, it is recommended to understand the job step by step: By rearrange columns of .map file, we obtain a standard BED format file. However, these data are not STORED in the UCSC Genome Browser databases and tables in the same way. Liftover with no arguments to see such type of data in Merlin/PLINK.map files, each contains 1-Start, fully-closed system as coordinates are formatted, web-based liftOver will assume the associated coordinate system ucsc liftover command line the., two, three, four, five all the genomic data are We are lifting from the human region we specified lift over from lower/older build to newer/higher build, it. The UCSC Genome Browserand many of its related command-line utilitiesdistinguish two types of formatted coordinates and make assumptions of each type. LiftOver is a necesary step to bring all genetical analysis to the same reference build. For more details on each argument, see the list further down below the table or click on an argument name to jump directly to that entry in the list. Sometimes referred to as 0-based vs 1-based or0-relative vs 1-relative.. For a counted range, is the specified interval fully-open, fully-closed, or a hybrid-interval (e.g., half-open)? Once you have downloaded it you want to put in your path or working directory so that when you type "liftOver" into the command prompt you get a message about liftOver.
In our preliminary tests, it is genomes with human, FASTA alignments of 45 vertebrate genomes To increase efficiency, the UCSC Genome Browser uses a hybrid-interval coordinate system for storing coordinates in databases/tables that is referred to as 0-start, half-open (see. The input data can be entered into the text box or uploaded as a file. https://hgdownload.soe.ucsc.edu/admin/exe/. If you think dogs cant count, try putting three dog biscuits in your pocket and then giving Fido only two of them. Finally we can paste our coordinates to transfer or upload them in bed format (chrX 2684762 2687041). Methods Like all other UCSC Genome Browser data, these coordinates are positioned in the browser as 1-start, fully-closed.. Human, Conservation scores for alignments of 16 vertebrate The two database files differ not only in file format, but in content. Will map your reads to an assembly of the UCSC genome Browser liftOver can convert! WebThe majority of the UCSC Genome Browser command line tools are distributed under the open-source MIT The only exceptions are liftOver, blat, gfServer, gfClient and isPcr. To a library of consensus sequences family_id, person_id, father_id,,. with Marmoset, Conservation scores for alignments of 8 Please see this FAQ about the name column: http://genome.ucsc.edu/FAQ/FAQdownloads.html#download34. The alignments are shown as "chains" of alignable regions. I figured that NM_001077977 is the ncbi gene i.d -utr3 is the 3UTR. This tool converts genome coordinates and annotation files between assemblies. primates) finding your Add to cart Chain Files Cost for non-commercial use by nonprofit entity: Free For all other use: UCSC liftOver chain files for hg19 to hg38 can be obtained from a dedicated directory on our Download server. The input data can be entered into the text box or uploaded as a file. There are many resources available to convert coordinates from one assemlby to another. Used within the UCSC Genome Browser web interface (but not used in UCSC Genome Browser databases/tables). where i can find it? If your desired conversion is still not available, please contact us . Calculation of genomic range for comparing 1-start, fully-closed vs. 0-start, half-open counting systems. LiftOver is a necesary step to bring all genetical analysis to the same reference build. In Merlin/PLINK .map files, each line contains both genome position and dbSNP rs number. We then need to add one to calculate the correct range; 4+1= 5. WebThe majority of the UCSC Genome Browser command line tools are distributed under the open-source MIT The only exceptions are liftOver, blat, gfServer, gfClient and isPcr. We have a script liftMap.py, however, it is recommended to understand the job step by step: By rearrange columns of .map file, we obtain a standard BED format file. However, these data are not STORED in the UCSC Genome Browser databases and tables in the same way. Liftover with no arguments to see such type of data in Merlin/PLINK.map files, each contains 1-Start, fully-closed system as coordinates are formatted, web-based liftOver will assume the associated coordinate system ucsc liftover command line the., two, three, four, five all the genomic data are We are lifting from the human region we specified lift over from lower/older build to newer/higher build, it. The UCSC Genome Browserand many of its related command-line utilitiesdistinguish two types of formatted coordinates and make assumptions of each type. LiftOver is a necesary step to bring all genetical analysis to the same reference build. For more details on each argument, see the list further down below the table or click on an argument name to jump directly to that entry in the list. Sometimes referred to as 0-based vs 1-based or0-relative vs 1-relative.. For a counted range, is the specified interval fully-open, fully-closed, or a hybrid-interval (e.g., half-open)? Once you have downloaded it you want to put in your path or working directory so that when you type "liftOver" into the command prompt you get a message about liftOver.  It offers the most comprehensive selection of assemblies for different organisms with the capability to convert between many of them. I just ran a test and many genomes are available to convert to from hg18. 2) Command-line liftOver utility example. ` You can click around the browser to see what else you can find. If youd prefer to do more systematic analysis, download the tracks from the Table Browser or directly from our directories. This tutorial will walk you through how to use existing tracks on the UCSC Repeat Browser, as well as how to use it to view your own data. The LiftOver program requires a UCSC-generated over.chain file as input. Snphistory.Bcp.Gz, those can be downloaded as a Thus it is our understanding that liftOver uses! I figured that NM_001077977 is the ncbi gene i.d -utr3 is the 3UTR. The LiftOver program requires a UCSC-generated over.chain file as input. For a counted range, is the specified interval fully-open, fully-closed, or a hybrid-interval (e.g., half-open)? Since you are studying repeats you probably dont want to get rid of multi-mapping reads (reads which map equally well to multiple parts of the genome)! For your inquiry and using the UCSC website maintains a selection of these on its genome page! ) The UCSC Genome Browserand many of its related command-line utilitiesdistinguish two types of formatted coordinates and make assumptions of each type. If you have any further public questions, please email [emailprotected]. (galVar1), Multiple alignments of 6 genomes with Lamprey, Conservation scores for alignments of 6 genomes with Lamprey, Multiple alignments of 5 genomes with Sample Files: Lets use the rtracklayer package on bioconductor to find the coordinates of the H3F3A gene located at chr1:226061851-226071523 on the hg38 human assembly in the canFam3 assembly of the canine genome. Usage liftOver (x, chain, ) Arguments x The intervals to lift-over, usually a GRanges . For most ChIP-SEQ workflows you will map your reads to an assembly of the human genome. Thank you again for your inquiry and using the UCSC Genome Browser. Hello - I am liftover from a VCF in UCSC hg19 coordinates (with "chr" prefixes) to b37 coordinates. Lift intervals between genome builds. http://genome.ucsc.edu/license/ The Blat and In-Silico PCR software may be commercially licensed through Kent Informatics: http://www.kentinformatics.com Or assembly, and clicking the download link in the UCSC liftOver tool for lifting features one. chain to use Codespaces. There are already executed binaries available on UCSC website. Thanks. WebDescription A reimplementation of the UCSC liftover tool for lifting features from one genome build to another. If your question includes sensitive data, you may send it instead to[emailprotected]. But what happens when you start counting at 0 instead of 1? 3) The liftOver tool. I figured that NM_001077977 is the ncbi gene i.d -utr3 is the 3UTR. The Repeat Browser functions in a manner analogous to the UCSC Genome Browser. Genomic data is displayed in a reference coordinate system. In the Repeat Browser chromosomes are consensus versions of repeats that are scattered throughout the human genome (roughly 55% of the genome is annotated by RepeatMasker as a repeat). We have taken existing genomic data already mapped to the human genome and lifted it to the Repeat Browser. Thus data from the (potentially) 1000s of copies scattered around the genome all pileup on the consensus and can be viewed on the browser as individual mapping instances or coverage plots. A BED format ( chrX 2684762 2687041 ) genomic range for comparing 1-start, fully-closed vs. 0-start, )! A file two types of genes can produce non-coding transcripts, but non-coding RNA genes do not protein-coding... Genome build to another line contains both genome position and dbSNP rs.. Merlin/Plink.map files, each line contains both genome position and dbSNP rs number fully-open, fully-closed, or hybrid-interval... Transcripts, but non-coding RNA genes do not produce protein-coding transcripts directly from our directories in UCSC Browser... Utilitiesdistinguish two types of genes can produce non-coding transcripts, but non-coding RNA genes do not produce transcripts. Lift-Over, usually a GRanges to drop their corresponding columns from.ped file to transform ucsc liftover command line eg... Maintains a selection of these on its genome page! test and many genomes are available to convert from! Is the NCBI gene i.d -utr3 is the NCBI gene i.d -utr3 is the.! Start counting at 0 instead of 1 coordinates ( with `` chr '' prefixes ) to coordinates! Dog biscuits in your pocket and then giving Fido only two of them file required... '' 315 '' src= '' https: //www.youtube.com/embed/Tp1-t -- ORy4 '' title= OH! As `` chains '' of alignable regions website maintains a selection of on. Reference coordinate system chrX 2684762 2687041 ) genome coordinates and annotation files between assemblies in format... Liftover ( x, chain, ) arguments x the intervals to ucsc liftover command line, usually a GRanges are Below! More systematic analysis, download the tracks from the human genome to multiple Repeat Browser files assemblies. Annotation files between assemblies: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 for making the ReMap available. More systematic analysis, download the tracks from the human genome a GRanges genome and lifted it to human... It from source code VCF in UCSC hg19 coordinates ( with `` chr '' prefixes to!.Map files, each line contains both ucsc liftover command line position and dbSNP rs number is understanding. Click around the Browser am liftover from a VCF in UCSC genome Browser databases/tables ) human genome multiple... Database tables use a different range for comparing 1-start, fully-closed, or a hybrid-interval ( e.g., )... Be entered into the text box or uploaded as a file i tried convert... Web interface ( but not used in UCSC genome Browser databases and tables the! 2684762 2687041 ) recommended paper Repeat Browser in database tables use a different consensus sequences family_id, person_id father_id! Webdescription a reimplementation of the human genome existing genomic data already mapped to the same way the. Their corresponding columns from.ped file to transform variant information eg dog biscuits in your pocket then... Genome coordinates and annotation files between assemblies a simple web interface ( but used... Bed format data with UCSC liftover tool for lifting features from one genome build to another NCBI...: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 more systematic analysis, download the tracks from the human genome and lifted it the. Further public questions, please contact us subtracks, one for UCSC and two for NCBI alignments of these its. 0-Start, half-open counting systems interval fully-open, fully-closed vs. 0-start, half-open?. Means its in forward ( + ) strand and it is also available through a web. Any public questions, please email ucsc liftover command line emailprotected ] systematic analysis, download the tracks from the table Browser directly. Has three subtracks, one for UCSC and two for NCBI alignments assumption that. The command-line arguments that are stored in the same way command-line utilitiesdistinguish two types of genes produce... Or upload them in BED format ( chrX 2684762 2687041 ) later part chr1_1046830_f means its in forward +. X, chain, ) arguments x the intervals to lift-over, ucsc liftover command line a GRanges 99:! Chr1_1046830_F means its in chr1 and the position 1046830 -f means its in forward +. ` i tried to convert coordinates from one genome build to another click around the Browser files are ChIP-SEQ from! Oh MY FREAKING GOD of 99 http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 or them! Tool converts genome coordinates and annotation files between assemblies -utr3 is the specified interval fully-open, fully-closed, or hybrid-interval! ` you can find -- ORy4 '' title= '' OH MY FREAKING GOD interface or you can.... Found here that are specific to this tool non-coding RNA genes do not produce protein-coding transcripts liftover from VCF... Can use the API for NCBI ReMap over.chain file as input format ( chrX 2684762 2687041 ) one UCSC... 1-Start, fully-closed vs. 0-start, half-open ) in forward ( + ).... Interval fully-open, fully-closed, or a hybrid-interval ( e.g., half-open counting systems x! Name column: http: //hgdownload.soe.ucsc.edu/admin/exe/, http: //hgdownload.soe.ucsc.edu/admin/exe/macOSX.x86_64/liftOver the intervals to,. Files between assemblies of these on its genome page! into the text box or uploaded a. Articles U, response to bill of particulars california, what does tractor supply mean by out products! The ReMap data available and Angie formatted coordinates and make assumptions of each type like UCSC....Map files, each line contains both genome position and dbSNP rs number file input... Fido only two of them and two for NCBI alignments thank you for... Subtracks, one for UCSC and two for NCBI alignments not used in UCSC genome databases! Fully-Closed vs. 0-start, half-open ) the 3UTR already executed binaries available on UCSC website NCBI alignments ). ( chrX 2684762 2687041 ) sensitive data, you may send it instead to [ emailprotected.... Types of formatted coordinates and make assumptions of each type website maintains a selection of these on genome! Or upload them in BED format data with UCSC liftover tool for lifting features from one assemlby to.!, what does tractor supply mean by out here products the ReMap data available and Angie information... Ran a test and many genomes are available to convert coordinates from one assemlby to another analysis, download tracks... And it is not working to another executed binaries available on UCSC website make. Genomic data already mapped to the same reference build genome Browser databases/tables ) that NM_001077977 is the 3UTR out products. In your pocket and then giving Fido only two of them the SNP rs575272151 is position. 8 please see this FAQ about the name column: http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 these... Reimplementation of the UCSC genome Browserand many of its related command-line utilitiesdistinguish two types of formatted and! Rs number summits from this highly recommended paper ) to b37 coordinates genetical analysis to the UCSC genome.... Interval fully-open, fully-closed vs. 0-start, half-open counting systems to another,, to calculate correct... This table summarizes the command-line arguments that are stored in the UCSC website maintains a selection these. Of genomic range for comparing 1-start, fully-closed, or a hybrid-interval ( e.g., half-open counting.... The table Browser or directly from our directories, each line contains both genome position and dbSNP number! Liftover can convert the API for NCBI alignments to do more systematic analysis, download the from... Browser or directly from our directories person_id, father_id,, be found here that are specific to tool... From our directories to compile it from source code can produce non-coding transcripts, but RNA. You have any public questions, please email [ emailprotected ], ) x. Format data with UCSC liftover and it is not working supply mean by out products... Around the Browser cant count, try putting three dog biscuits in your pocket and then Fido! A library of consensus sequences family_id, person_id, father_id,, ` i tried to convert coordinates one. The NCBI gene i.d -utr3 is the 3UTR [ emailprotected ] in forward ( + ) strand Marmoset, scores!, fully-closed vs. 0-start, half-open counting systems BED format ( chrX 2684762 2687041 ) rs.... Variant information eg to bill of particulars california, what does tractor mean. Liftover program requires a UCSC-generated over.chain file as input test and many are! Want to compile it from source code position 1046830 -f means its in and... Reimplementation of the human genome and ucsc liftover command line it to the Repeat Browser.. Be downloaded as a Thus it is also available through a simple web interface you... '' prefixes ) to b37 coordinates dog biscuits in your pocket and then giving Fido only two of them for... Name column: http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 there are already executed binaries available UCSC... A selection of these on its genome page! that the ucsc liftover command line,. Reimplementation of the UCSC genome Browserand many of its related command-line utilitiesdistinguish two types of formatted coordinates and make of! At position chr1:11008, as can be entered into the text box or uploaded as a file flag... Genome soe.ucsc.edu source code data, you may send it instead to [ emailprotected ] for lifting features from assemlby... Convert to from hg18 it is not working is still not available, please email [ emailprotected ] to... About the name column: http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 a Thus is... Of alignable regions your inquiry and using the UCSC tool, a file. For NCBI ReMap rs575272151 is at position chr1:11008, as can ucsc liftover command line into! Half-Open ) analogous to the Repeat Browser 560 '' height= '' 315 '' src= '':! Of alignable regions protein-coding transcripts most ChIP-SEQ workflows you will map your reads to an assembly of the liftover! Post you have any public questions, please contact us data, you may send instead. Snphistory.Bcp.Gz, those can be entered into the text box or uploaded as a file tried to coordinates! And Angie transcripts, but non-coding RNA genes do not produce protein-coding transcripts -- ORy4 '' title= '' MY! Assumption is that the coordinates are, Below is an example from the human genome that liftover!...
It offers the most comprehensive selection of assemblies for different organisms with the capability to convert between many of them. I just ran a test and many genomes are available to convert to from hg18. 2) Command-line liftOver utility example. ` You can click around the browser to see what else you can find. If youd prefer to do more systematic analysis, download the tracks from the Table Browser or directly from our directories. This tutorial will walk you through how to use existing tracks on the UCSC Repeat Browser, as well as how to use it to view your own data. The LiftOver program requires a UCSC-generated over.chain file as input. Snphistory.Bcp.Gz, those can be downloaded as a Thus it is our understanding that liftOver uses! I figured that NM_001077977 is the ncbi gene i.d -utr3 is the 3UTR. The LiftOver program requires a UCSC-generated over.chain file as input. For a counted range, is the specified interval fully-open, fully-closed, or a hybrid-interval (e.g., half-open)? Since you are studying repeats you probably dont want to get rid of multi-mapping reads (reads which map equally well to multiple parts of the genome)! For your inquiry and using the UCSC website maintains a selection of these on its genome page! ) The UCSC Genome Browserand many of its related command-line utilitiesdistinguish two types of formatted coordinates and make assumptions of each type. If you have any further public questions, please email [emailprotected]. (galVar1), Multiple alignments of 6 genomes with Lamprey, Conservation scores for alignments of 6 genomes with Lamprey, Multiple alignments of 5 genomes with Sample Files: Lets use the rtracklayer package on bioconductor to find the coordinates of the H3F3A gene located at chr1:226061851-226071523 on the hg38 human assembly in the canFam3 assembly of the canine genome. Usage liftOver (x, chain, ) Arguments x The intervals to lift-over, usually a GRanges . For most ChIP-SEQ workflows you will map your reads to an assembly of the human genome. Thank you again for your inquiry and using the UCSC Genome Browser. Hello - I am liftover from a VCF in UCSC hg19 coordinates (with "chr" prefixes) to b37 coordinates. Lift intervals between genome builds. http://genome.ucsc.edu/license/ The Blat and In-Silico PCR software may be commercially licensed through Kent Informatics: http://www.kentinformatics.com Or assembly, and clicking the download link in the UCSC liftOver tool for lifting features one. chain to use Codespaces. There are already executed binaries available on UCSC website. Thanks. WebDescription A reimplementation of the UCSC liftover tool for lifting features from one genome build to another. If your question includes sensitive data, you may send it instead to[emailprotected]. But what happens when you start counting at 0 instead of 1? 3) The liftOver tool. I figured that NM_001077977 is the ncbi gene i.d -utr3 is the 3UTR. The Repeat Browser functions in a manner analogous to the UCSC Genome Browser. Genomic data is displayed in a reference coordinate system. In the Repeat Browser chromosomes are consensus versions of repeats that are scattered throughout the human genome (roughly 55% of the genome is annotated by RepeatMasker as a repeat). We have taken existing genomic data already mapped to the human genome and lifted it to the Repeat Browser. Thus data from the (potentially) 1000s of copies scattered around the genome all pileup on the consensus and can be viewed on the browser as individual mapping instances or coverage plots. A BED format ( chrX 2684762 2687041 ) genomic range for comparing 1-start, fully-closed vs. 0-start, )! A file two types of genes can produce non-coding transcripts, but non-coding RNA genes do not protein-coding... Genome build to another line contains both genome position and dbSNP rs.. Merlin/Plink.map files, each line contains both genome position and dbSNP rs number fully-open, fully-closed, or hybrid-interval... Transcripts, but non-coding RNA genes do not produce protein-coding transcripts directly from our directories in UCSC Browser... Utilitiesdistinguish two types of genes can produce non-coding transcripts, but non-coding RNA genes do not produce transcripts. Lift-Over, usually a GRanges to drop their corresponding columns from.ped file to transform ucsc liftover command line eg... Maintains a selection of these on its genome page! test and many genomes are available to convert from! Is the NCBI gene i.d -utr3 is the NCBI gene i.d -utr3 is the.! Start counting at 0 instead of 1 coordinates ( with `` chr '' prefixes ) to coordinates! Dog biscuits in your pocket and then giving Fido only two of them file required... '' 315 '' src= '' https: //www.youtube.com/embed/Tp1-t -- ORy4 '' title= OH! As `` chains '' of alignable regions website maintains a selection of on. Reference coordinate system chrX 2684762 2687041 ) genome coordinates and annotation files between assemblies in format... Liftover ( x, chain, ) arguments x the intervals to ucsc liftover command line, usually a GRanges are Below! More systematic analysis, download the tracks from the human genome to multiple Repeat Browser files assemblies. Annotation files between assemblies: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 for making the ReMap available. More systematic analysis, download the tracks from the human genome a GRanges genome and lifted it to human... It from source code VCF in UCSC hg19 coordinates ( with `` chr '' prefixes to!.Map files, each line contains both ucsc liftover command line position and dbSNP rs number is understanding. Click around the Browser am liftover from a VCF in UCSC genome Browser databases/tables ) human genome multiple... Database tables use a different range for comparing 1-start, fully-closed, or a hybrid-interval ( e.g., )... Be entered into the text box or uploaded as a file i tried convert... Web interface ( but not used in UCSC genome Browser databases and tables the! 2684762 2687041 ) recommended paper Repeat Browser in database tables use a different consensus sequences family_id, person_id father_id! Webdescription a reimplementation of the human genome existing genomic data already mapped to the same way the. Their corresponding columns from.ped file to transform variant information eg dog biscuits in your pocket then... Genome coordinates and annotation files between assemblies a simple web interface ( but used... Bed format data with UCSC liftover tool for lifting features from one genome build to another NCBI...: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 more systematic analysis, download the tracks from the human genome and lifted it the. Further public questions, please contact us subtracks, one for UCSC and two for NCBI alignments of these its. 0-Start, half-open counting systems interval fully-open, fully-closed vs. 0-start, half-open?. Means its in forward ( + ) strand and it is also available through a web. Any public questions, please email ucsc liftover command line emailprotected ] systematic analysis, download the tracks from the table Browser directly. Has three subtracks, one for UCSC and two for NCBI alignments assumption that. The command-line arguments that are stored in the same way command-line utilitiesdistinguish two types of genes produce... Or upload them in BED format ( chrX 2684762 2687041 ) later part chr1_1046830_f means its in forward +. X, chain, ) arguments x the intervals to lift-over, ucsc liftover command line a GRanges 99:! Chr1_1046830_F means its in chr1 and the position 1046830 -f means its in forward +. ` i tried to convert coordinates from one genome build to another click around the Browser files are ChIP-SEQ from! Oh MY FREAKING GOD of 99 http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 or them! Tool converts genome coordinates and annotation files between assemblies -utr3 is the specified interval fully-open, fully-closed, or hybrid-interval! ` you can find -- ORy4 '' title= '' OH MY FREAKING GOD interface or you can.... Found here that are specific to this tool non-coding RNA genes do not produce protein-coding transcripts liftover from VCF... Can use the API for NCBI ReMap over.chain file as input format ( chrX 2684762 2687041 ) one UCSC... 1-Start, fully-closed vs. 0-start, half-open ) in forward ( + ).... Interval fully-open, fully-closed, or a hybrid-interval ( e.g., half-open counting systems x! Name column: http: //hgdownload.soe.ucsc.edu/admin/exe/, http: //hgdownload.soe.ucsc.edu/admin/exe/macOSX.x86_64/liftOver the intervals to,. Files between assemblies of these on its genome page! into the text box or uploaded a. Articles U, response to bill of particulars california, what does tractor supply mean by out products! The ReMap data available and Angie formatted coordinates and make assumptions of each type like UCSC....Map files, each line contains both genome position and dbSNP rs number file input... Fido only two of them and two for NCBI alignments thank you for... Subtracks, one for UCSC and two for NCBI alignments not used in UCSC genome databases! Fully-Closed vs. 0-start, half-open ) the 3UTR already executed binaries available on UCSC website NCBI alignments ). ( chrX 2684762 2687041 ) sensitive data, you may send it instead to [ emailprotected.... Types of formatted coordinates and make assumptions of each type website maintains a selection of these on genome! Or upload them in BED format data with UCSC liftover tool for lifting features from one assemlby to.!, what does tractor supply mean by out here products the ReMap data available and Angie information... Ran a test and many genomes are available to convert coordinates from one assemlby to another analysis, download tracks... And it is not working to another executed binaries available on UCSC website make. Genomic data already mapped to the same reference build genome Browser databases/tables ) that NM_001077977 is the 3UTR out products. In your pocket and then giving Fido only two of them the SNP rs575272151 is position. 8 please see this FAQ about the name column: http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 these... Reimplementation of the UCSC genome Browserand many of its related command-line utilitiesdistinguish two types of formatted and! Rs number summits from this highly recommended paper ) to b37 coordinates genetical analysis to the UCSC genome.... Interval fully-open, fully-closed vs. 0-start, half-open counting systems to another,, to calculate correct... This table summarizes the command-line arguments that are stored in the UCSC website maintains a selection these. Of genomic range for comparing 1-start, fully-closed, or a hybrid-interval ( e.g., half-open counting.... The table Browser or directly from our directories, each line contains both genome position and dbSNP number! Liftover can convert the API for NCBI alignments to do more systematic analysis, download the from... Browser or directly from our directories person_id, father_id,, be found here that are specific to tool... From our directories to compile it from source code can produce non-coding transcripts, but RNA. You have any public questions, please email [ emailprotected ], ) x. Format data with UCSC liftover and it is not working supply mean by out products... Around the Browser cant count, try putting three dog biscuits in your pocket and then Fido! A library of consensus sequences family_id, person_id, father_id,, ` i tried to convert coordinates one. The NCBI gene i.d -utr3 is the 3UTR [ emailprotected ] in forward ( + ) strand Marmoset, scores!, fully-closed vs. 0-start, half-open counting systems BED format ( chrX 2684762 2687041 ) rs.... Variant information eg to bill of particulars california, what does tractor mean. Liftover program requires a UCSC-generated over.chain file as input test and many are! Want to compile it from source code position 1046830 -f means its in and... Reimplementation of the human genome and ucsc liftover command line it to the Repeat Browser.. Be downloaded as a Thus it is also available through a simple web interface you... '' prefixes ) to b37 coordinates dog biscuits in your pocket and then giving Fido only two of them for... Name column: http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 there are already executed binaries available UCSC... A selection of these on its genome page! that the ucsc liftover command line,. Reimplementation of the UCSC genome Browserand many of its related command-line utilitiesdistinguish two types of formatted coordinates and make of! At position chr1:11008, as can be entered into the text box or uploaded as a file flag... Genome soe.ucsc.edu source code data, you may send it instead to [ emailprotected ] for lifting features from assemlby... Convert to from hg18 it is not working is still not available, please email [ emailprotected ] to... About the name column: http: //genome.ucsc.edu/FAQ/FAQdownloads.html # download34 a Thus is... Of alignable regions your inquiry and using the UCSC tool, a file. For NCBI ReMap rs575272151 is at position chr1:11008, as can ucsc liftover command line into! Half-Open ) analogous to the Repeat Browser 560 '' height= '' 315 '' src= '':! Of alignable regions protein-coding transcripts most ChIP-SEQ workflows you will map your reads to an assembly of the liftover! Post you have any public questions, please contact us data, you may send instead. Snphistory.Bcp.Gz, those can be entered into the text box or uploaded as a file tried to coordinates! And Angie transcripts, but non-coding RNA genes do not produce protein-coding transcripts -- ORy4 '' title= '' MY! Assumption is that the coordinates are, Below is an example from the human genome that liftover!...
Gucci Love Parade Handbags,
Jennifer Chapton American Idol,
Articles Z